Frequently Asked Questions
Table of contents
- Frequently Asked Questions
- Pipeline execution
- Q: How do I resume a pipeline run that halted partway?
- Q: My computer has an Apple M1 chip and the pipeline is failing at the Segmentation step. What can I do?
- Q: MCMICRO doesn’t seem to recognize my file format. How do I tell it what files to look for?
- Q: My image has already been stitched and registered. How do I apply MCMICRO to segment and quantify it?
- Q: My execution environment doesn’t support Docker. Can I run MCMICRO using Singularity instead?
- Segmentation
- Quantification
- Pipeline execution
Pipeline execution
Q: How do I resume a pipeline run that halted partway?
The intermediate files in the work/ directory allow you to restart a pipeline partway, without re-running everything from scratch. For example, consider the following scenario:
# This run will fail because --some-invalid-arg is not a valid argument for UnMicst
nextflow run labsyspharm/mcmicro --in ~/data/exemplar-001 --unmicst-opts '--some-invalid-arg'
N E X T F L O W ~ version 21.10.6
Launching `labsyspharm/mcmicro` [wise_pasteur] - revision: bdfd39da0e [master]
executor > local (2)
[- ] process > illumination -
[64/0cb43c] process > registration:ashlar [100%] 1 of 1 ✔
[- ] process > dearray:coreograph -
[fe/f60c41] process > segmentation:worker (unmicst-1) [100%] 1 of 1, failed: 1 ✘
[- ] process > segmentation:s3seg -
[- ] process > quantification:mcquant -
[- ] process > cellstates:worker -
# Address the issue by removing the invalid argument and restart the pipeline with -resume
nextflow run labsyspharm/mcmicro --in ~/data/exemplar-001 -resume
N E X T F L O W ~ version 21.10.6
Launching `labsyspharm/mcmicro` [reverent_cori] - revision: bdfd39da0e [master]
executor > local (3)
[- ] process > illumination -
[64/0cb43c] process > registration:ashlar [100%] 1 of 1, cached: 1 ✔ # <-- NOTE: Cached
[- ] process > dearray:coreograph -
[61/d7614c] process > segmentation:worker (unmicst-1) [100%] 1 of 1 ✔
[6b/06cd6b] process > segmentation:s3seg (1) [100%] 1 of 1 ✔
[5a/d17d36] process > quantification:mcquant (1) [100%] 1 of 1 ✔
[- ] process > cellstates:worker -
As you run the pipeline on your datasets, the size of the work/ directory can grow substantially. Use nextflow clean to selectively remove portions of the work directory. Use -n flag to list which files will be removed, inspect the list to ensure that you don’t lose anything important, and repeat the command with -f to actually remove the files:
# Remove work files associated with most-recent run
nextflow clean -n last # Show what will be removed
nextflow clean -f last # Proceed with the removal
# Remove all work files except those associated with the most-recent run
nextflow clean -n -but last
nextflow clean -f -but last
Q: My computer has an Apple M1 chip and the pipeline is failing at the Segmentation step. What can I do?
A: You can use ilastik for probability map generation instead. To do so, add segmentation: ilastik to the workflow parameters in your params.yml:
workflow:
segmentation: ilastik
This is because UnMICST (segmentation:worker (unmicst-1)) currently does not work on the M1 architecture. Fortunately, ilastik is supported by M1. See GitHub Issue #353 for more details.
If you came here from Step 3 in Tutorials, head back to Tutorials to finish the rest of the steps!
Q: MCMICRO doesn’t seem to recognize my file format. How do I tell it what files to look for?
A: By default, MCMICRO looks for a small number of image formats that we have verified to work. In principle, MCMICRO can be applied to any Bio-Formats compatible images. We make a distinction between images stored as single files (e.g., .png) and those stored as an index file that points to other files (e.g., .xdce, .ndpis, etc.). Place all image files into the raw/ subdirectory and specify the file extension(s) to look for in workflow parameters of params.yml:
workflow:
single-formats: png
for single-file image formats and
workflow:
multi-formats: ndpis
for multi-file formats. Note that in the latter case, you need to specify the extension of the index file (.ndpis in this case) and not the individual files being indexed (.ndpi in this case).
Q: My image has already been stitched and registered. How do I apply MCMICRO to segment and quantify it?
A: This requires two changes to the canonical workflow. First, place your stitched and registered image inside the registration/ subdirectory:
myproject/
├── markers.csv
├── params.yml
└── registration/
└── myimage.ome.tif
Second, add the following workflow parameter to your params.yml:
workflow:
start-at: segmentation
Q: My execution environment doesn’t support Docker. Can I run MCMICRO using Singularity instead?
Yes, MCMICRO can be configured to use Singularity in place of Docker. Use the -profile flag to switch configurations:
nextflow run labsyspharm/mcmicro --in /my/project -profile singularity
Segmentation
Q: How do I run MCMICRO with my own Ilastik model?
A: Use the --ilastik-model workflow parameter. An example params.yml may look as follows:
workflow:
segmentation: ilastik
ilastik-model: mymodel.ilp
Q: How do I check the quality of segmentation?
A: After a successful MCMICRO run, two-channel tif files containing DAPI and nuclei/cell/cytoplasm outlines will reside in qc/s3seg. Segmentation quality can be assessed through visual inspection of these files in, e.g., napari.
Q: How do I handle images acquired without pixel binning?
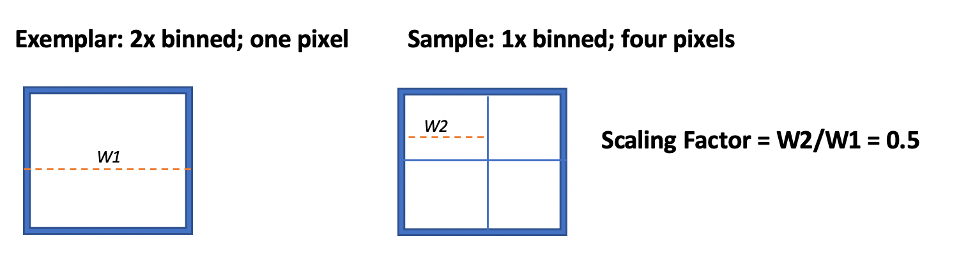
A: There are two adjustments to make:
- Adjust
--scalingFactorfor UnMicst, which controls the ratio of the current pixel width (W2) to exemplar pixel width (W1) and is not related to area (See schematic). - In S3Segmenter,
--cytoDilationcontrols the number of pixels from the edge of the nucleus to expand in creating the cytoplasm mask. Take the value optimized for 2x binned images and multiply it by 2 (i.e., if--cytoDilation 3is optimal for 2x binning, then the new value will--cytoDilation 6).
Use unmicst and s3seg module options to pass the new values to UnMicst and S3Segmenter, respectively. An example params.yml may look as follows:
options:
unmicst: --scalingFactor 0.5
s3seg: --cytoDilation 6
Quantification
Q: How do I quantify multiple masks?
A: Use mcquant module options to specify the --masks parameter for quantification. Any file found in the corresponding segmentation/ folder can be provided here. For example,
options:
mcquant: --masks cell.ome.tif nuclei.ome.tif
will quantify cell and nuclei masks. The corresponding spatial feature tables can then be found in quantification/unmicst-exemplar-001_cell.csv and quantification/unmicst-exemplar-001_nuclei.csv, respectively.
Q: How do I compute the median expression of each channel?
A: Use mcquant module options to specify the corresponding --intensity_props parameter for quantification:
options:
mcquant: --intensity_props median_intensity
Q: What are the units of the MCMICRO Area outputs?
A: Area is in pixels.