MCMICRO tutorial
Here we show an example of how to execute MCMICRO on two exemplar datasets using the command line (Nextflow) interface on a local computer.
Note: Nextflow is compatible with Linux or Mac (through the terminal), but Windows users will need to set up a Windows Subsystem for Linux or use the Galaxy workflow.
If you are not running MCMICRO on your local computer, please head to Platforms for platform-specific instructions.
Following this tutorial can verify that MCMICRO is correctly installed and running on your computer by demonstrating the expected inputs and outputs for the exemplar datasets.
Step 0: Be sure to install nextflow and Docker before proceeding through these steps.
Enter the commands nextflow run hello and docker run hello-world to verify that both Nexflow and Docker* are functional.
*If using a Windows environment, you may need to run Docker as Administrator.
*If using a MacOS environment, you may need to increase the amount of RAM allocated to Docker.
Step 1: Ensure you have the latest version of the pipeline
nextflow pull labsyspharm/mcmicro
Step 2: Download exemplar data.
Replace /my/path with where you want to store the files. (Use . for path to download files into the current directory.)
# Download exemplar-001
nextflow run labsyspharm/mcmicro/exemplar.nf --name exemplar-001 --path /my/path
# Download exemplar-002
nextflow run labsyspharm/mcmicro/exemplar.nf --name exemplar-002 --path /my/path
Both exemplar datasets have the directory structure of
exemplar-001 or exemplar-002
├── markers.csv
├── raw/
| ├── exemplar-001-cycle-06.ome
| ├── exemplar-001-cycle-07.ome
| └── ...
|
└── illumination/
├── ...
├── exemplar-001-cycle-06-dfp.tif
├── exemplar-001-cycle-06-ffp.tif
├── ...
└── ...
NOTE: For this tutorial, we demonstrate running the pipeline with the input of raw images and illumination profiles (in illumination). To see a more comprehensive description of inputs for MCMICRO, please see the Input section under Inputs/Outputs.
By default, exemplar-001 contains cycles 6 through 8, while exemplar-002 contains all 10 cycles. Use nextflow run labsyspharm/mcmicro/exemplar.nf --help to learn how to download a different range of cycles for each exemplar.
Each cycle has one .ome.tif file in raw/ and two .tif files in illumination/. These images look like the following:

exemplar-001-cycle-06.ome.tif
This is the first image in a stack of 24 images showing the Hoechst stain for DNA.

exemplar-001-cycle-06-dfp.tif
This is the first image of a stack of 4 images, previewed in Fiji with auto adjustments made to brightness and contrast. The image contains the dark-field illumination profile.

exemplar-001-cycle-06-ffp.tif
This is the first image of a stack of 4 images, previewed in Fiji with auto adjustments made to brightness and contrast. The image contains the flat-field illumination profile.
Step 3: Use --in to point the pipeline at the data.
In the commands below, /my/path should match what was used to download exemplar data with exemplar.nf command above (. can again be used to point to the current directory).
# Run the pipeline on exemplar data (starting from the registration step, by default)
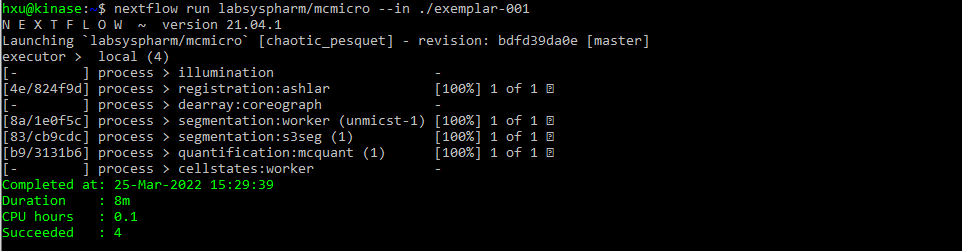
nextflow run labsyspharm/mcmicro --in /my/path/exemplar-001
# params.yml in exemplar-002 will tell MCMICRO to treat data as a tissue microarray
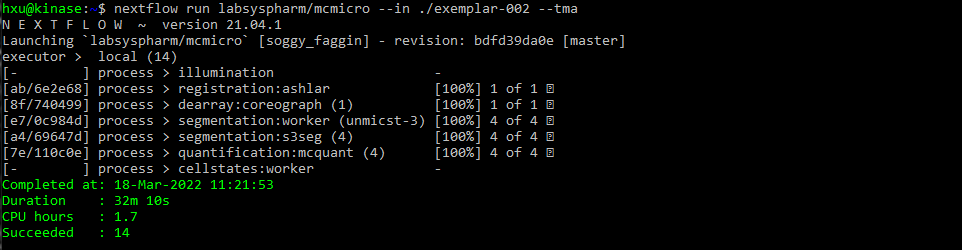
nextflow run labsyspharm/mcmicro --in /my/path/exemplar-002
If your computer has an Apple M1 chip, you may need to specify ilastik for probability maps at this step. Read more on the FAQ page.
*On an average workstation, it takes approximately 5-10 minutes to process exemplar-001 from start to finish. Exemplar-002 is substantially larger, and takes 30-40 minutes on an average workstation.
After a successful run, the following text will be displayed for exemplar-001 and exemplar-002:
The pipeline will generate the following directory, depending on the modules used.
Expand to see exemplar-001 and exemplar-002 output files*
*raw/ and illumination/ contents will remain the same.
exemplar-001
├── markers.csv
├── raw/
├── illumination/
├── registration/
| └── exemplar-001.ome
├── probability-maps/
| └── unmicst/
| └── exemplar-001-pmap.tif
├── segmentation/
| ├── cell.ome
| └── nuclei.ome
├── quantification/
| └── unmicst-exemplar-001_cell.csv
└── qc/
├── provenance/
| ├── quantification·mcquant(1).txt
| ├── quantification·mcquant(1).sh
| ├── reigstration·ashlar.txt
| ├── registration·ashlar.sh
| ├── segmentation·s3seg(1).txt
| ├── segmentation·s3seg(1).sh
| ├── segmentation·worker(unmicst-1).txt
| └── segmentation·workder(unmicst-1).sh
├── s3seg/
| └── unmicst-exemplar-001/
| ├── cellOutlines.ome
| └── nucleiOutlines.ome
├── unmicst/
| └── exemplar-001-Preview_1.tif
└── params.yml
exemplar-002
├── markers.csv
├── raw/
├── illumination/
├── registration/
| └── exemplar-002.ome
├── dearray/
| ├── masks/
| | ├── 1_mask.tif
| | ├── 2_mask.tif
| | ├── 3_mask.tif
| | └── 4_mask.tif
| ├── 1.tif
| ├── 2.tif
| ├── 3.tif
| └── 4.tif
├── probability-maps/
| └── unmicst/
| ├── 1-pmap.tif
| ├── 2_pmap.tif
| ├── 3_pmap.tif
| └── 4_pmap.tif
├── segmentation/
| ├── unmicst-1/
| | ├── cell.ome
| | └── nuclei.ome
| ├── unmicst-2/
| | ├── cell.ome
| | └── nuclei.ome
| ├── unmicst-3/
| | ├── cell.ome
| | └── nuclei.ome
| └── unmicst-4/
| ├── cell.ome
| └── nuclei.ome
├── quantification/
| ├── unmicst-1_cell.csv
| ├── unmicst-2_cell.csv
| ├── unmicst-3_cell.csv
| └── unmicst-4_cell.csv
└── qc/
├──coreo/
| ├── centroidsY-X.txt
| └── TMA_MAP.tif
├── provenance/
| ├── dearraycoreograph(1).txt
| ├── dearraycoreograph(1).sh
| ├── quantification·mcquant(1).txt
| ├── quantification·mcquant(1).sh
| ├── ...
| ├── quantification·mcquant(4).txt
| ├── quantification·mcquant(4).sh
| ├── reigstration·ashlar.txt
| ├── registration·ashlar.sh
| ├── segmentation·s3seg(1).txt
| ├── segmentation·s3seg(1).sh
| ├── ...
| ├── segmentation·s3seg(4).txt
| ├── segmentation·s3seg(4).sh
| ├── segmentation·worker(unmicst-1).txt
| ├── segmentation·worker(unmicst-1).sh
| ├── ...
| ├── segmentation·worker(unmicst-4).txt
| └── segmentation·workder(unmicst-4).sh
├── s3seg/
| └── unmicst-exemplar-001/
| ├── cellOutlines.ome
| └── nucleiOutlines.ome
├── unmicst/
| └── exemplar-001-Preview_1.tif
└── params.yml
# Working with TMA array (tma:true in params.yml) produces the dearray/ directory
Step 4: (Recommended) Visual inspection of quality control (qc/) files
Depending on the modules used, directories coreo/, unmicst/ and s3seg/ may contain .tif or .ome.tif images for inspection.
Here we demonstrate visual inspection using Fiji (Download Fiji here). You can use any image viewing/processing software that works for .ome.tif and .tif files.
s3seg/
Check that
cellOutlines.ome.tifandnucleiOutlines.ome.tifshow satisfactorily outlined areascellOutlines.ome.tiffound underqc/s3seg1/unmicst-exemplar-001/can be previewed as Hyperstack in Fiji. Each cycle appears as a 2-image stack.You can split stack into individual images. Then, choose Image>Color>Merge Channels to overlay outline with raw image for visual inspection.
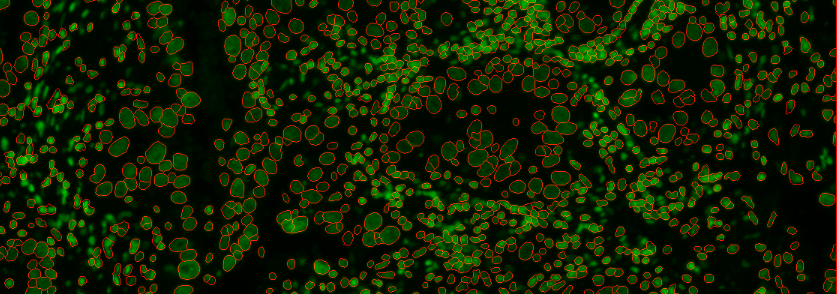
cellOutlines.ome.tifCell outlines overlaid with raw image, zoomed in on an arbitrary area. (This example shows the first available cycle (Cycle 6) in the default exemplar-001 data set .)Read Parameter Tuning for S3Segmenter for common troubleshooting scenarios.
coreo/
Check for correct partitioning of TMAs
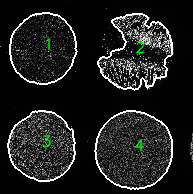
TMA_MAP.tifexemplar-002Read Parameter Tuning for Coreograph for common troubleshooting scenarios.
unmicst/
Examination of
qc/unmicst/is generally not necessary ifqc/s3seg/outlines are satisfactoryHowever, if segmentation results found in
qc/s3seg/are not desirable, UnMICSTqcfiles can provide a clue for what went wrong.Read Parameter Tuning for UnMICST for common troubleshooting scenarios.
More details on output files and quality control can be found in Inputs/Outputs.